
Peer Reviewed
Nicolaides-Baraitser Syndrome
AUTHORS:
Trevor Smith • Peggy Hsuanyu Kuo • Mariam Fahim, DO
AFFILIATIONS:
Western University College of Osteopathic Medicine of the Pacific, Pomona, California
CITATION:
Smith T, Kuo PH, Fahim M. Nicolaides-Baraitser syndrome. Consultant. 2021;61(6):e19-e21. doi:10.25270/con.2020.10.00024
Received May 2, 2020. Accepted August 10, 2020. Published online October 20, 2020.
DISCLOSURES:
The authors report no relevant financial relationships.
CORRESPONDENCE:
Mariam Fahim, DO, Associate Professor of Pediatrics, Western University College of Osteopathic Medicine of the Pacific, 309 E Second St, Pomona, CA 91766-1854 (mfahim@westernu.edu)
A 4½-year-old boy who previously had been diagnosed with Williams syndrome (WS) presented to a pediatric clinic after the family had immigrated to the United States from Jalisco, Mexico.
History. The boy had been born at 40 weeks of gestation via spontaneous vaginal delivery with no complications. His birth weight was 2.9 kg. There was no family history of developmental delay or congenital defects. The boy’s developmental milestones were delayed, including his first steps not taken until age 2 years, an absence of speech, and a failure to toilet train by age 4 years. In addition, he had primary incontinence requiring diapers at all times, poor social interactions with other children, and failure to thrive.
Physical examination. A neurologist was consulted, and a complete evaluation was performed. On physical examination, the patient presented with thin hair, a smooth philtrum, puffy eyes, and slightly anteverted nares (Figure 1). He was able to make steady eye contact and maintained a social smile at his mother and the examiner. Furthermore, the patient was able to make babbling noises but could not form words. He had low muscle tone throughout the upper and lower extremities, with normal muscle strength. His gait was steady with shoulder-width base. The rest of the examination findings were within normal limits. From the assessment, the neurologist agreed that the patient most likely had William syndrome but recommended that the patient be seen by a geneticist due to the complete absence of speech and language development.
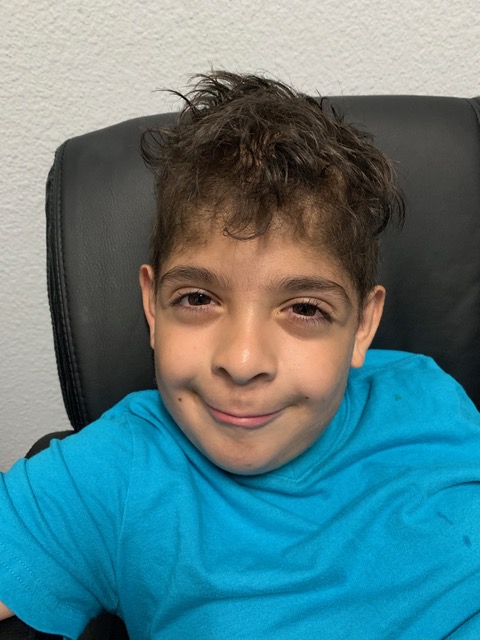
Figure 1. The patient showed physical characteristics of smooth philtrum, puffy eyes, and slightly anteverted nares.
Differential diagnosis. Although the patient displayed the typical features of WS, such as long philtrum and intellectual disability, he did not exhibit the strength in spoken language and hypercalcemia as seen in other individuals with WS.1 Other differential diagnoses included Coffin-Siris syndrome (CSS) and Nicolaides-Baraitser syndrome (NCBRS). CSS produces similar coarse facial features as seen in the patient, but it is also classically characterized by absent or hypoplastic fifth nails or distal phalanx of the hands and/or feet,1 and these features were not present in the patient. Common features characteristic of individuals with NCBRS include sparse scalp hair, prominence of interphalangeal joints, coarse facies, and intellectual disability,1 all of which were exhibited by the patient.
Geneticist’s examination. Upon examination by the geneticist, the patient’s weight, height, and head circumference were reported to be in the 65th, 59th, and 36th percentiles, respectively. In addition to the sparse scalp hair, the geneticist noted the narrow forehead and hypertrichosis on the sides and posterior midline of cranium (Figure 2).
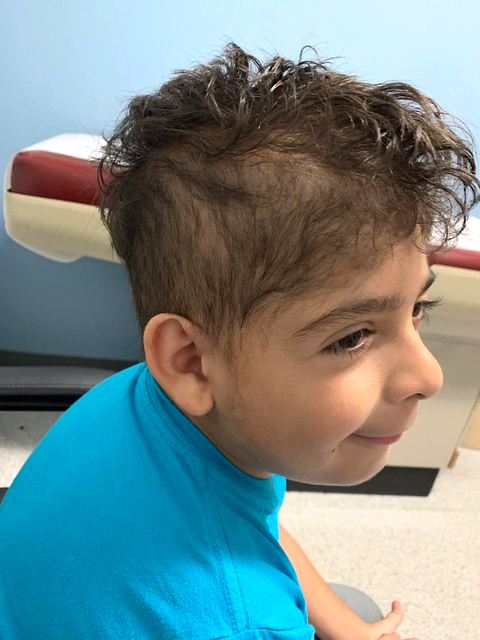
Figure 2. The patient exhibited sparse scalp hair, a narrow forehead, and localized hypertrichosis on posterior midline of the cranium.
He had down-slanting palpebral fissures, long eye lashes, dark circles around the eyes, an inner canthal distance of 3 cm, and a left palpebral fissure distance of 2.7 cm. The patient’s ears were normal in shape and position. His nose had a very broad base and was anteverted, and his columella was low-hanging. His philtrum was very long and smooth, and he had a thin upper lip without a Cupid’s bow.
The joints in his hands were hyperextensible with prominent interphalangeal joints and decreased subcutaneous fat. The fourth and fifth metacarpals were short. The fingers were thin, and the distal phalanges were broadened in the hands and feet (Figures 3 and 4). All other examination findings were within normal limits.
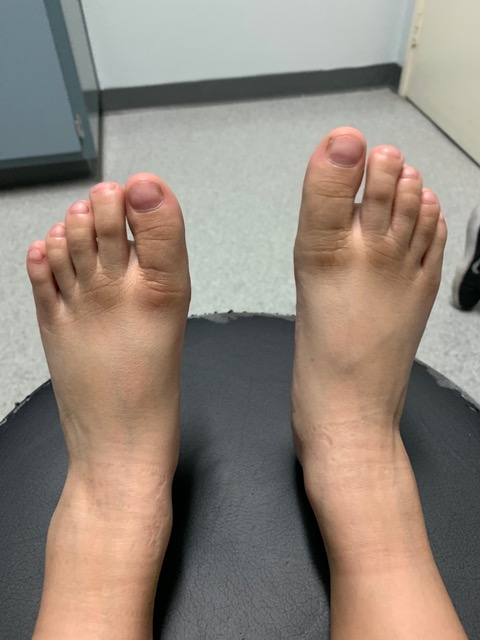
Figure 3. Brachydactyly of the patient’s feet, moderately prominent interphalangeal joints, and broad distal phalanges.
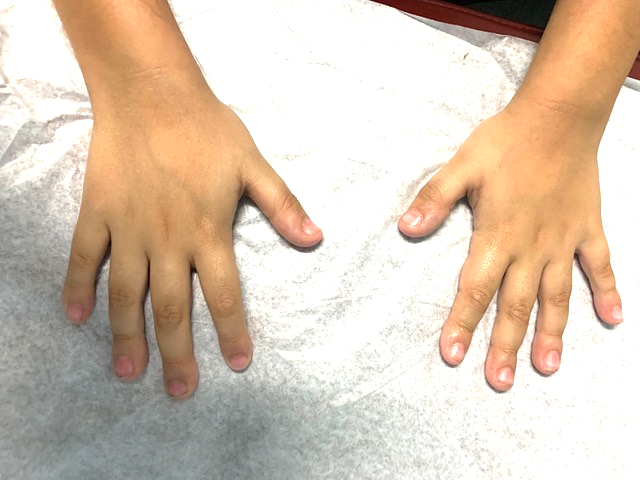
Figure 4. The patient had slightly prominent interphalangeal joints with broad distal phalanges of hands.
Multigene sequencing was ordered, which located a de novo heterozygous deletion of exons 15 to 19 in SMARCA2 on 9p24.3—the only gene known to cause NCBRS. The patient was confirmed to have NCBRS at this point, as evidenced by the dysmorphic features and SMARCA2 deletion sequence variant.
Management and follow-up. The patient’s mother was counseled on the characteristic traits of NCBRS, as well as the genetic basis and autosomal dominant inheritance pattern of de novo sequence variations in SMARCA2. Recommendations were made to the mother for an electroencephalography (EEG) evaluation to determine whether seizures were present, since they are associated with NCBRS. In addition, echocardiography was ordered to detect any congenital heart conditions, which have been reported in some individuals with NCBRS. Furthermore, the patient was encouraged to begin occupational therapy, speech therapy, and/or physical therapy as needed when starting preschool. Most individuals with NCBRS remain nonverbal and require additional support in communication and education.
At his most recent follow-up visit, the boy was 8 years old, and his height, weight, and body mass index were in the 27th, 85th, and 95th percentiles, respectively. EEG showed normal results, and echocardiography showed a small patent foramen ovale with normal left ventricular size and systolic shortening, as well as a normal left ventricular ejection fraction of 65%. He continues to have absence of speech and severe developmental delays, and he works with a multidisciplinary team that includes speech, behavioral, and occupational therapists. To date, he has not had any known episodes of seizures or regression. He is growing well but will require supportive therapies throughout his life.
Discussion. NCBRS is a rare genetic disease that was first identified in 1993 by pediatric neurologist Paola Nicolaides and geneticist Michael Baraitser.2 NCBRS is caused by sequence variations in SMARCA2 that arise de novo.3 SMARCA2 encodes the catalytic subunit of a member of the SWI/SNF chromatin remodeling complex, which has important implications in gene expression, differentiation, and development.3 Exons 15 to 19 of SMARCA2 specifically correspond to the helicase ATP-binding sequence motif of the SNF2 ATPase domain, and sequence variations in this region disrupt the DNA and ATP-binding that are important for cell differentiation and transcriptional regulation via chromatin remodeling.4
Individuals with NCBRS can have a wide array of symptoms with varying severity. The most common symptoms are intellectual disability, sparse hair, prominent interphalangeal joints, coarse facies, microcephaly, and seizures.1 To date, approximately 100 cases have been described in the literature. Due to its extreme rarity and similarities to numerous other genetic diseases such as WS and CSS,5 a diagnosis of NCBRS is difficult to make and is often missed. The diagnosis often requires multiple workups that encompass various tests and medical specialties. Early diagnosis of this condition is especially important due to the progressive nature of its severity.
REFERENCES:
- Abdul-Rahman O. Nicolaides-Baraitser syndrome. GeneReviews. October 15, 2015. Accessed September 8, 2020. https://www.ncbi.nlm.nih.gov/books/NBK321516/
- Sousa SB, Abdul-Rahman OA, Bottani A, et al. Nicolaides–Baraitser syndrome: delineation of the phenotype. Am J Med Genet A. 2009;149A(8):1628-1640. doi:10.1002/ajmg.a.32956
- Van Houdt JK, Nowakowska BA, Sousa SB, et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44(4):445-S1. doi:10.1038/ng.1105
- Sousa SB, Hennekam RC; Nicolaides-Baraitser Syndrome International Consortium. Phenotype and genotype in Nicolaides-Baraitser syndrome. Am J Med Genet C Semin Med Genet. 2014;166C(3):302-314. doi:10.1002/ajmg.c.3140
- Wieczorek D, Bögershausen N, Beleggia F, et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22(25):5121-5135. doi:10.1093/hmg/ddt366